
Gene Regulation and RNA Systems Biology

What we do.
We aim to understand the regulation of gene expression in its full complexity using molecular, systems and cell biological tools in human cells and C. elegans.
RNA is at the center of this regulatory complexity; every aspect of its lifecycle can be regulated, from its production and processing to its intracellular localization and degradation.
Through our research we aim to answer questions such as:
· How are the instructions, when and how a gene is expressed, encoded in the DNA?
· How are the many gene regulatory mechanisms coordinated to ensure proper functioning of the cell?
· How do genetic mutations change normal processing and ultimately lead to disease?

Who we are.
The Mikl Lab is located at the Department of Human Biology of the University of Haifa, Israel. It was established in fall 2020 and strives to be a diverse, inclusive and interdisciplinary group of scientists.
We are driven by scientific curiosity as well as the conviction that elucidating the basic principles of biology is the foundation for understanding disease states and developing new therapeutic strategies.
Selected Publications

Martin Mikl, Davide Eletto, Minkyoung Lee, Atefeh Lafzi, Farah Mhamedi, Simona Baghai Sain, Kristina Handler, Andreas E. Moor. Nucleic Acids Research 50(18), 10643–10664 (2022).
Intracellular localization of mRNAs is crucial for functional compartmentalization of many cells such as neurons, but we still lack a systematic understanding of how the transcript sorting machinery works in a sequence-specific manner. Here we performed the first massively parallel reporter assay for RNA localization in neurons. This revealed how the localization potential is encoded in the 3’UTR sequence. By combining high-throughput assays, biochemistry and bioinformatics we obtained novel mechanistic insights into this important regulatory mechanism and identified novel players and sequence elements involved in mediating mRNA dendritic targeting or soma restriction.

High-throughput interrogation of programmed ribosomal frameshifting in human cells
Martin Mikl, Yitzhak Pilpel, Eran Segal. Nature Communications 11, 3061 (2020).
Programmed ribosomal frameshifting (PRF) is a strikingly robust and precise process by which the ribosome shifts reading frame in a defined fraction of translation events, leading to the production of two different proteins from the same mRNA. This process is most prominent in viruses like HIV and SARS coronavirus, where it is a widespread and indispensable mechanism to regulate the production of key enzymes. Here, we developed a massively parallel reporter assay that allows for accurate high-throughput quantification of ribosomal frameshifting in human cells. We combined the power of fluorescent frameshifting reporters with rational design of DNA sequences, which allowed us to systematically decipher determinants of PRF efficiency across frameshifting events and assay natural variation in HIV gag-pol frameshifting, providing the first systematic large-scale investigation of ribosomal frameshifting.
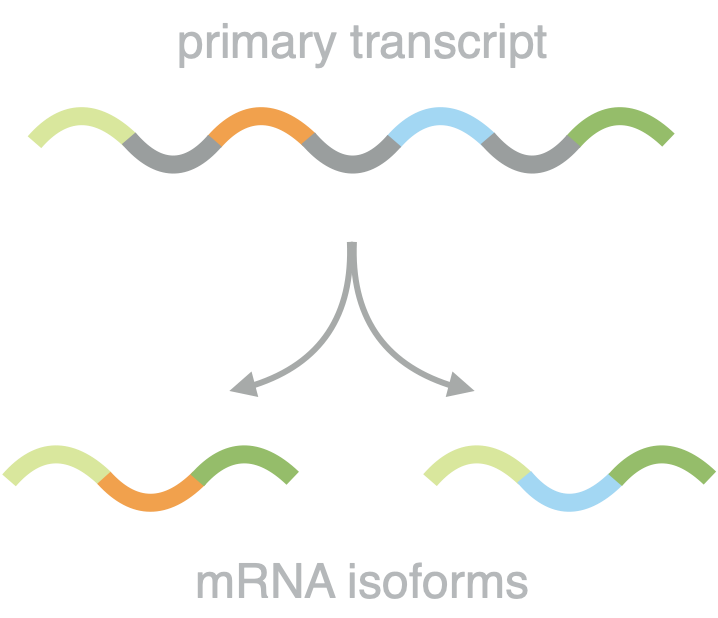
Dissecting splicing decisions and cell-to-cell variability with designed sequence libraries
Martin Mikl, Amit Hamburg, Yitzhak Pilpel, Eran Segal. Nature Communications 10, 4572 (2019).
Pre-mRNA splicing is influenced by a multitude of factors, ranging from local sequence motifs to the greater genomic environment. This complexity has up to now hindered a comprehensive understanding of the individual contributions to the splicing decision in native sequence contexts. In this study, we overcame these limitations by combining the power of massively parallel reporter assays and rationally designed sequence libraries, while maintaining expression from a defined and constant genomic location. This approach allowed us to reduce the regulatory complexity and pinpoint causative sequence changes. We survey different splicing types in a comprehensive and comparative way and develop a predictive model for splice site usage. By quantifying the relative abundance of protein isoforms we follow splicing decisions to the level of the final functional gene product and for the first time assess the cell-to-cell variability of splicing in large scale.